
Tools
GRMapp
Authors: Glunčić M, Paar V, Vlahović I, Mršić L- GRMapp.jar - Download GRM application
- Input case file - Download input case file (human chromosome19 sequence, RefSeq: NC_000019.10)
- Output TRs report case file - Download output TR report (.csv file) with list of all TRs in sequence
- Output TRs sequences file - Download output TRs sequences file (.txt file) with all TRs sequences
What is GRMapp?
- GRMapp is GUI application for identification of tandem repeats in genomic sequence. It is very fast, efficient and simple, with powerful graphical user interface. It doesn’t require any prior knowledge of sequences structure and doesn't require any user defined parameters or thresholds. It is robust to all types of mutations in repeat copies and can identify tandem repeats with various complexities in the sequence pattern.
- GRMapp GUI (JAR file) can be run on any platform which have Java Runtime Environment (JRE) installed (freely available at Java SE Runtime Environment 8 Downloads).
How input file looks like?
- Curent version suports .fasta, .fa and .txt filesor any similar format with the header (the name and/or sequence description) first line, followed by a body. The body should consist of genomic sequence which can be broken into lines (like in fasta file) or can also be in one line. The sequence should generally consist of four nucleotides, A, C, G, T and the symbol ‘N’ on possible places where the nucleotides have not yet been identified. All other symbols within the sequence that may appear as a typo or ambiguity between two or more nucleotides are converted during the load process into ‘N’s.
How to start?
-
Click on the 'Start' button in main window and load the genomic sequence:
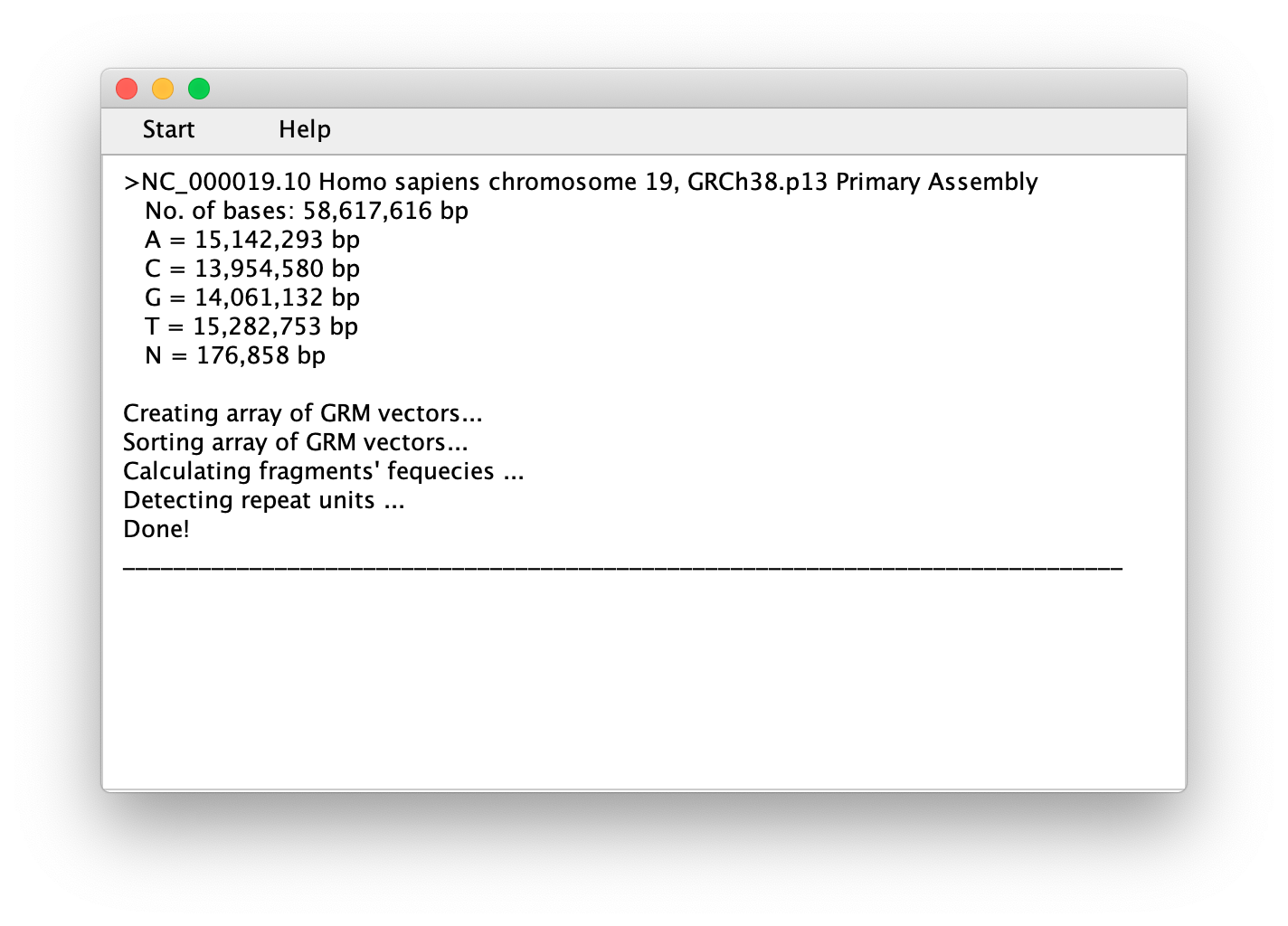
-
After the sequence loading phase, the application autonomously goes through the steps described in the Algorithm outline and, as a result, delivers a report listing all the TRs in the observed sequence:
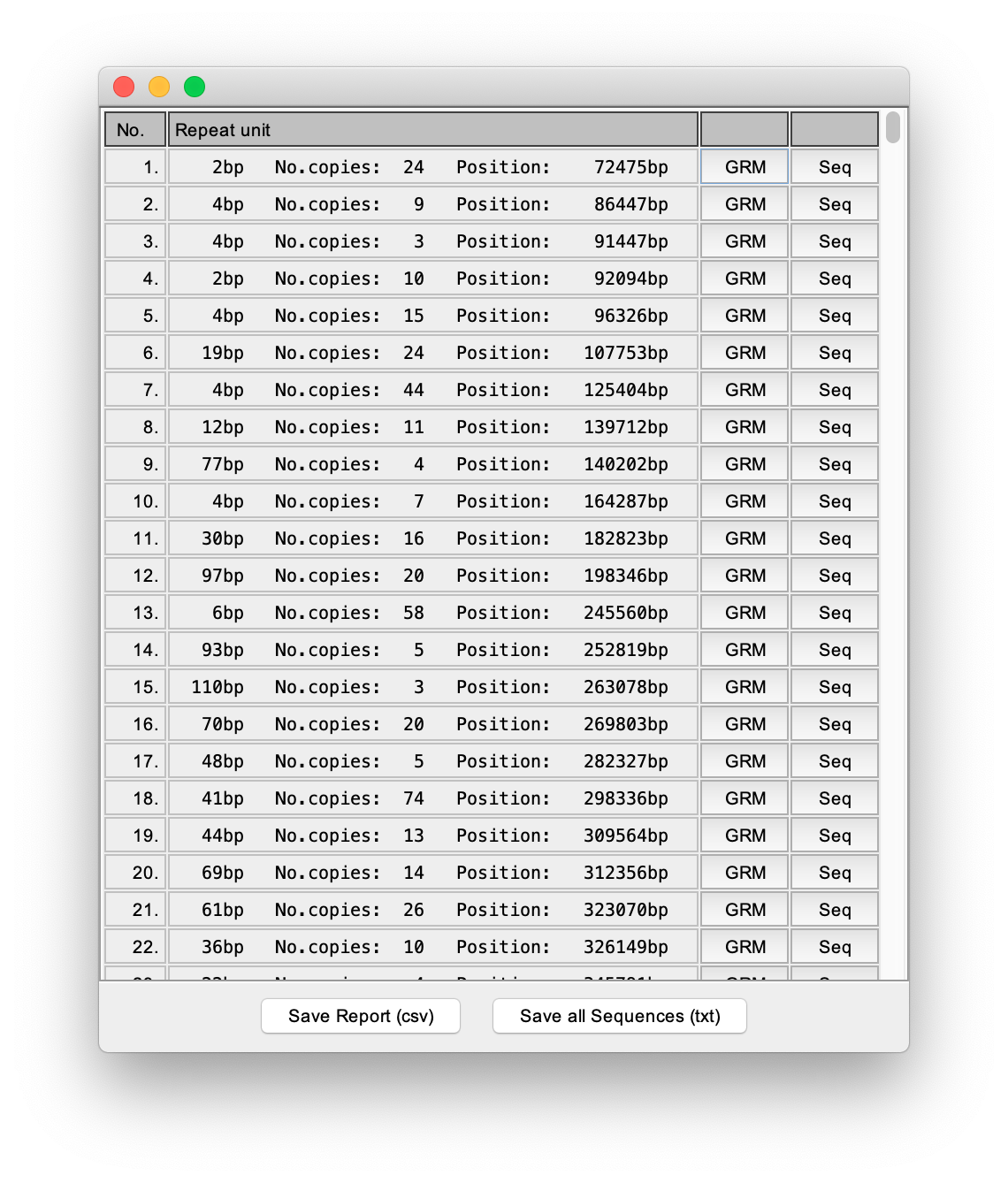
At the bottom of the main report there are two buttons, the button for exporting the entire main report, and the button for exporting all TRs sequences in one file.
-
In the report, next to each identified TR, there is the button that displays the GRM diagram of the subarray in which TR was found:
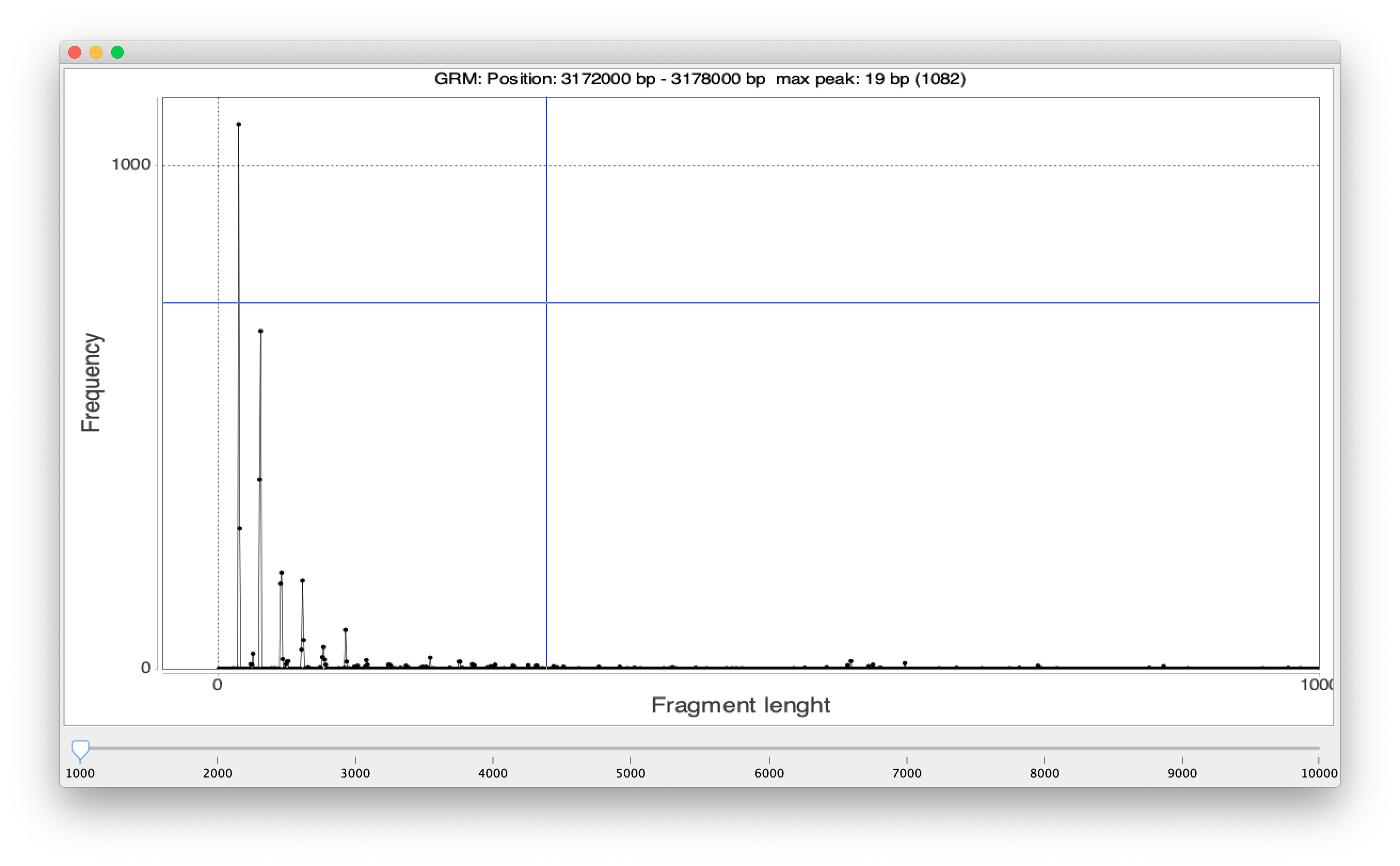
-
In the report, next to each identified TR, there is the button that displays new report with the repetitive sequence itself. In Sequence report, the sequence is broken by a dominant key string obtained in the last step described in Algorithm outline. In each sequence report, each nucleotide is colored with its own color to make it easier visual identification of deviations from the ideal TR copies, such as deletions, insertions or substitution of individual or groups of nucleotides. The exception are very long sequences of TRs that require a lot of time for coloring and therefore they are displayed without colors.
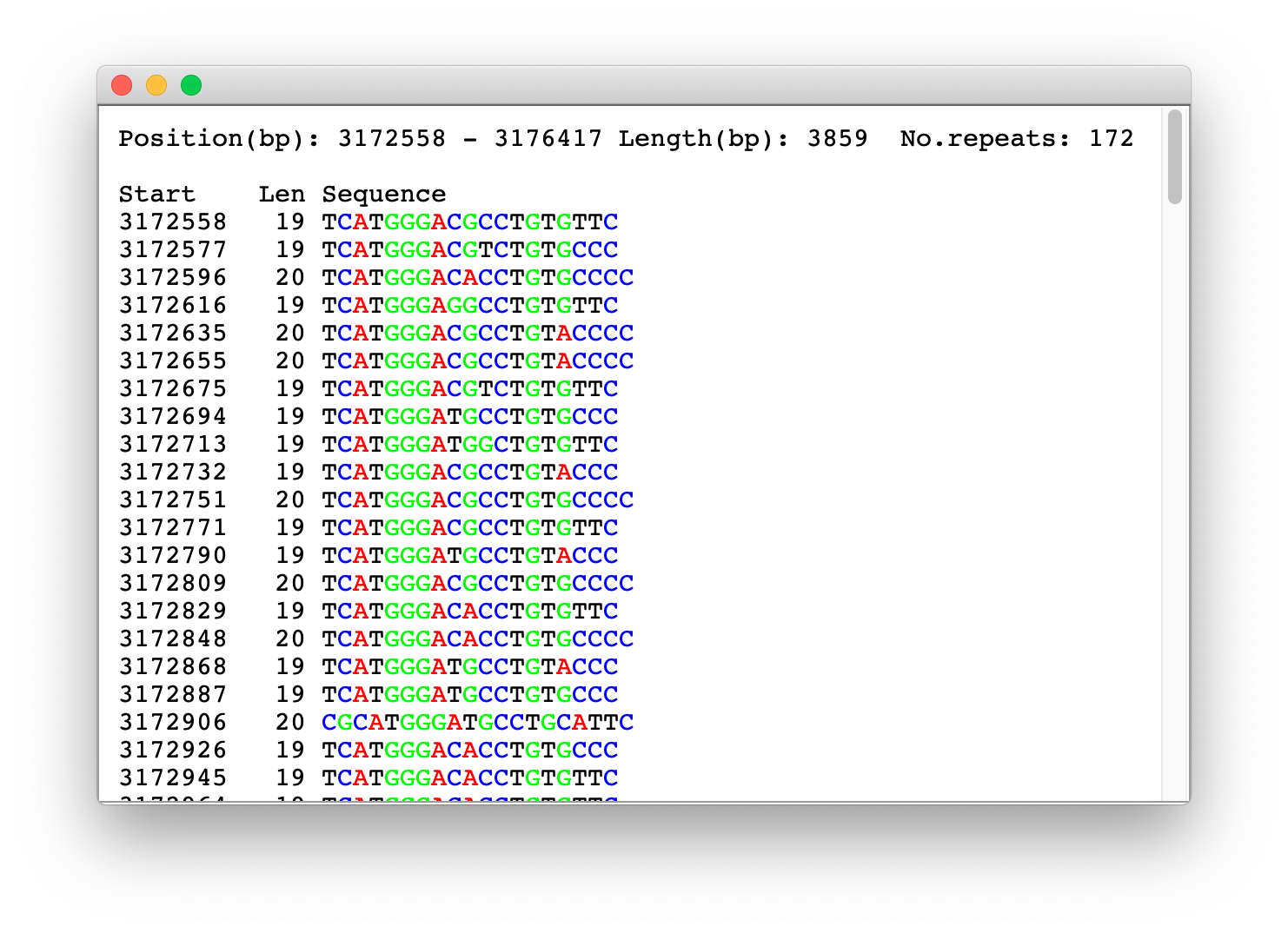
ALPHAsub
- Authors: Glunčić M
What is ALPHAsub?
- ALPHAsub is a tool for finding all alpha monomers in genomic sequence.
How input file looks like?
- Current version suports regular fasta (.fa or.fasta) files with fasta header (e.g. ">chr_name").
How to start?
- This Linux program needs only a single fasta (.fa or.fasta) file as input. The aplication starts with: ./ALPHAasub.exe fasta_file_name.fa. Results are automatically saved to the folder where original input file is located.
- ALPHAsub.exe Download
GRManalytics
- Authors: Glunčić M, Jerković H, Mršić L
What is GRManalytics?
- GRManalytics is a set of tools for analyzing the results of GRMapp and ALPHAsub applications. The results of the analysis are dot matrix, consessus sequence, CENP-B boxes, suprachromosomal families and HOR structures. .
How input file looks like?
- Input file(s) depends of programs' flags. Program runs in command line and file names are passed as an argument in the command line.
How to start?
-
Program may need admin privileges to be able to write out data.
Download and extract files in some folder (eg. C:\New Folder\).
Open command prompt (Start->Run->cmd.exe press Enter). In command prompt change current folder to the one where files are been exctracted (eg. cd "c:\New folder")
Start the calculation by passing flag, file path and location as an argument to the program(eg. grmanalytics.exe -s "C:\New Folder\file.fasta").
Advanced: Additional argument can be passed to the program to change parameter (eg. grmanalytics.exe -d "C:\New Folder\file01.fasta" "C:\New Folder\file02.fasta").
Available parameters:
parameter |
description |
|
|---|---|---|
| -d file01_name file02_name | finding distance between two fasta sequences | |
| -m file_name | finding dot matrix for alpha ensemble | |
| -b file_name | finding CENP-B boxes in alpha ensemble | |
| -s file_name | finding suprachomosomal families for alpha ensemble | |
| -h file_name | finding HOR structures for alpha ensemble |
- Possible file_name_with_TR_ensemble formats:
- TR_name (string) TR_length (int) TR_unit_sequence (string)
- TR_name (string) TR_direction (string) TR_position (int) TR_length (int) TR_unit_sequence (string)
- TR_name (string) TR_direction (string) TR_position (int) TR_consensus_difference (string) TR_length (int) TR_unit_sequence (string)
How output looks like:
- Outputs are stored in .txt and/or .rtf file(s) (by default txt only). Files are saved in folder where input file is located, and are named: OriginalFileName_type_of_analyses.txt
- GRManalytics.exe Download
CLT_Find
- Authors: Vlahović I
What is CLT_Find?
- CLT_Find is a tool for calculating trinucleotide frequencies in genomic sequence.
How input file looks like?
- Current version suports .txt files; without any headers, just bare nucleotide sequence.
How to start?
- This Windows program needs only a single .txt file as input. Results are automatically saved to the folder where original input file is located.
- CLT_Find.zip Download




